Евсеев М.А.
Обсуждение побочных эффектов наиболее востребованной в современной клинической практике группы лекарственных средств – нестероидныхпротивовоспалительных препаратов (НПВП) – традиционно ограничивается дискуссией о потенциальной (по мнению терапевтов) или реальной (по мнению хирургов) опасности токсического влияния данных препаратов на слизистую пищеварительного тракта. Однако, как пищеварительный тракт не органичен слизистой оболочкой желудка и кишечника, так и спектр побочных эффектов традиционных НПВП со стороны органов пищеварения не исчерпывается лишь их гастротоксичностью. Не менее значимой проблемой, связанной с широким применением НПВП, является их гепатотоксический потенциал. Гепатотоксичность НПВП, не представляясь большинству современных исследователей как некое исключение из правила или артефакт, сегодня служит предметом пристального изучения уже на уровне национальных департаментов здравоохранения [6,13,22,27]. На первый взгляд, мишени токсического воздействия НПВП различны – слизистая оболочка пищеварительной трубки и печеночная паренхима. Однако говорить о разнонаправленном побочном действии данной группы препаратов было бы слишком упрощенно. Представляется, что гастро– и гепатотоксичность НПВП являются звеньями одной патогенетической цепи, следствием которой может быть возникновение массивной геморрагии из верхнего отдела пищеварительного тракта. Указанное осложнение находится уже в компетенции хирургов, что определяет возможность изложения концепции патогенетической взаимосвязи гастро– и гепатотоксичности НПВП представителю данной специальности.
Ключевыми моментами данной патогенетической модели являются: прямое токсическое влияние НПВП на интактную или патологически измененную печеночную паренхиму с возникновением острой или усугублением хронической печеночной недостаточности, портальная гастропатия у больных с циррозом печени и синдромом портальной гипертензии, НПВП–индуцированное повреждение слизистой гастродуоденальной зоны.
Прямое токсическое действие НПВП на печень. Лекарственное поражение печени, в том числе и НПВП, является одной из серьезных проблем современной гепатологии. Это обусловлено не столько частотой возникновения нежелательных реакций, сколько большой вероятностью неблагоприятных исходов в случае их возникновения: до 25% всех случаев фульминантной печеночной недостаточности связано именно с лекарственным поражением печени. НПВП, являясь с химической точки зрения крайне неоднородной группой лекарственных препаратов, оказывают сходное терапевтическое действие и вызывают однотипные побочные эффекты. N. O’Connor et al. (2003) на основании изучения эпидемиологических аспектов гепатотоксичности нестероидных препаратов сделали вывод о том, что почти все современные НПВП качественно обладают гепатотоксичностью, количественно выраженной в большей или меньшей степени. Несмотря на то что относительный риск клинически значимого повреждения печени вследствие применения НПВП относительно невысок (8–27 случаев на 100000 пациентов в год), последствия возникшего НПВП–индуцированного повреждения печени зачастую становятся самыми серьезными: фульминантная печеночная недостаточность и гепаторенальный синдром [35]. Указанные нежелательные побочные эффекты терапии НПВП, следствием которых в конечном итоге явились гибель пациентов или необходимость экстренных трансплантаций печени, послужили причиной запрещения на использование в клинической практике беноксапрофена, дроксикама, бромфенака, пирпрофена, фенклофенака и в ряде государств нимесулида [10,11,15,17,21–23,31,33,36,38–40,42,45].
Считается, что повреждение печеночной паренхимы вследствие приема НПВП может возникать в различный временной промежуток: непосредственно после начала терапии, в процессе двух–трехнедельного курса лечения, спустя недели или даже месяцы после его завершения. P. Aithal et al. (1999) полагают, что наиболее часто клинико–лабораторные проявления НПВП–индуцированного повреждения печени находятся в интервале 6–12 недель с момента начала терапии. I. Lacroix et al. (2004), анализируя особенности клинической манифестации острых повреждений печени, связанных с приемом НПВП, отмечает, что у 5–27% пациентов такие повреждения протекают бессимптомно и могут быть выявлены лишь по повышению активности трансаминаз плазмы [9,21]. Большинство исследователей подчеркивает, что риск лекарственного поражения печени возрастает при наличии хронического диффузного заболевания печени любой этиологии. При этом нарушение метаболизма лекарств прямо пропорционально выраженности хронической печеночноклеточной недостаточности [4,22,35,41,48].
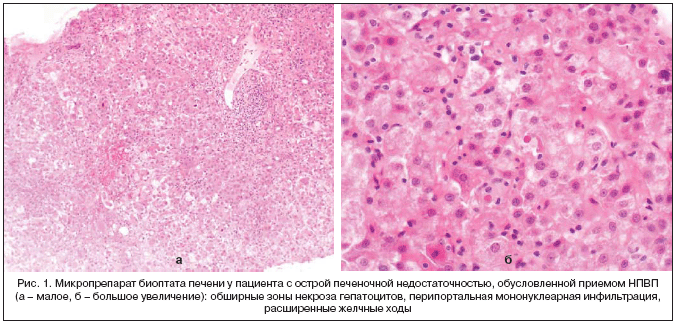
Как указывают H. Tan et al. (2007), клинико–морфологические формы токсического воздействия НПВП на печень весьма вариабельны: острый гепатит, холестаз, холестатический гепатит, имеющий тенденцию к хронизации гранулематозный гепатит, хронический холестаз с дуктопенией, желтая атрофия печени, проявляющаяся фульминантной печеночной недостаточностью. Авторы полагают, что наиболее характерной особенностью морфологической картины при НПВП–индуцированном повреждении печени является сочетание обширного гепатоцеллюлярного некроза с перипортальной мононуклеарной инфильтрацией и холестатическим компонентом (рис. 1) [48].
Несмотря на немалое количество исследований, посвященных проблеме НПВП–индуцированной гепатотоксичности, собственно механизм повреждения печеночной паренхимы на клеточном и субклеточном уровнях сегодня остается предметом дискуссии [5].
N. O’Connor et al. (2003) приводит две наиболее часто упоминающиеся в литературе причины гепатотоксичности НПВП: 1) реакции гиперчувствительности, 2) метаболическое повреждение (аберрация). О возможности НПВП–индуцированного повреждения печени, являющегося следствием реакций гиперчувствительности (аллергических реакций), по мнению R. Greaves et al. (2001), свидетельствует факт развития «перекрестной» токсичности при разделенных во времени курсах терапии химически сходными препаратами (диклофенак – тиапрофеновая кислота), а также факт клинического проявления гепатотоксичности у препарата при повторном курсе терапии [20,34]. R. Bort et al. (1999) на основании обобщения экспериментальных данных представили последовательность событий при повреждении гепатоцита НПВП следующим образом. Основной мишенью НПВП на субклеточном уровне являются митохондрии. В процессе метаболизма НПВП цитохромом Р450 образуются производные НПВП (например, 5–гидроксидиклофенак и n,5–дигидроксидиклофенак), которые способны влиять на процессы переноса электронов в дыхательной цепи на кристах митохондрий, приводя к нарушению окислительного фосфорилирования, синтеза АТФ и энергетическому дефициту в клетке. Не исключено, что токсическим влиянием на митохондрии обладают и нативные НПВП. Нарушение процессов окислительного фосфорилирования в митохондриях и микросомальное окисление некоторых НПВП (например, напроксена) приводят к активации свободно–радикального окисления, перекисному окислению липидов мембран, НАДФ и тиоловых групп белков, результатом чего, в конечном счете, является дезорганизация мембран, гибель гепатоцита и синтопичных клеточных структур (клетки желчных протоков) [14,26–30]. Возможно, что в процессе дезорганизации цитолемма приобретает антигенные свойства, что приводит к индукции аутоиммунного ответа и морфологически проявляется как перипортальный отек и момонуклеарная инфильтрация.
Типичным примером гепатотоксического действия НПВП может служить представленный A. Schattner et al. (2000) случай развития острой печеночной недостаточности и гепаторенального синдрома, этиологически связанный с приемом нимесулида [45].
70–летняя пациентка была госпитализирована по поводу выраженной слабости и клинически манифестированной желтухи, развившихся за 2 суток до госпитализации. Из объективных симптомов в момент госпитализации были выявлены желтушность кожи и иктеричность склер, тахикардия (117 в мин.). Больная страдала ожирением и остеоартритом коленных суставов, по поводу которого в течение нескольких лет принимала диклофенак. Впоследствии диклофенак был заменен нимесулидом (100 мг дважды в сутки), 5–дневный курс терапии которым был завершен за две недели до настоящей госпитализации. Лабораторные показатели на момент госпитализации: гемоглобин 135 г/л, лейкоциты 16х109/л (лейкоцитарная формула не изменена), тромбоциты 203х109/л, СОЭ 28 мм/ч, азот мочевины 5,8 ммоль/л, креатинин плазмы 70,7 мкмоль/л, электролиты плазмы и биохимический анализ мочи (за исключением желчных пигментов) в пределах нормы; общий билирубин плазмы 216 мкмоль/л, алкалин фосфатаза 285 МЕ/л, ?–ГТП 173 МЕ/л, АсАТ 1700 МЕ/л, АлАТ 1240 МЕ/л, ЛДГ 1460 МЕ/л, ?–амилаза сыворотки 285 МЕ/л, общий холестерол 3,9 ммоль/л, триглицериды 1,6 ммоль/л; альбумин сыворотки 25 г/л, глобулины 40 г/л; протромбиновое время 21с, МНО 2,3, АЧТВ 33 с (впоследствии не изменялось при внутривенном введении витамина К). Серологические пробы на вирусные гепатиты А, В, С и Е, вирус простого герпеса, ВИЧ, ПЦР на ДНК HBV и РНК HCV отрицательные. Инфекционная этиология заболевания была исключена, аутоантитела не выявлены, скрининг–тесты на экзогенные токсические агенты отрицательные. Биохимический анализ крови, проведенный за месяц до госпитализации, отклонений от нормы не выявил. Выполненные непосредственно после госпитализации ультразвуковое исследование и компьютерная томография брюшной полости отклонений от нормы не выявили. При гистологическом исследовании ткани печени, полученной при пункционной тонкоигольной биопсии на 5–й день нахождения в стационаре, были выявлены массивные некрозы гепатоцитов с выраженной смешанной (плазматические клетки, лимфоциты, эозинофилы и гранулоциты) воспалительной инфильтрацией портальных трактов и паренхимы. Данная картина согласовывалась с клиническими признаками фульминантного гепатита. К концу первой недели пребывания в стационаре отмечено нарастание желтухи с повышением общего билирубина до 484 мкмоль/л (максимальный общий билирубин 564 мкмоль/л) при снижении уровня печеночных ферментов более чем наполовину от их первоначальных значений, отмечено внезапное развитие гипогликемии. На четвертый день нахождения в стационаре у пациентки выявлено повышение азота мочевины до 10,8 ммоль/л и креатинина сыворотки до 124 мкмоль/л с последующим нарастанием к концу первой недели до 177 мкмоль/л. Это сопровождалось возникновением олигурии (300 мл/сут.) при неизмененном осадке мочи, FENa 0,1%, отсутствием ответа на водную нагрузку. Проведение терапии глюкокортикоидами (метилпреднизолон внутривенно 125 мг/сут.), назначение урсодезоксихолевой кислоты, лактулозы, диета с низким содержанием белка, впоследствии – инфузии глюкозы и допамина видимого эффекта не оказали. Прогрессировали гипоальбуминемия, коагулопатия, нарушения ментального статуса. Дважды отмечалась мелена. При дальнейшем нарастании почечной недостаточности (повышение креатинина плазмы до 495 мкмоль/л, лейкоцитурия, микрогематурия, FENa 2%) дважды проводились сеансы гемодиализа. Произведен посев крови на стерильность, назначены антибиотики широкого спектра действия. Однако состояние пациентки ухудшалось, нарастали расстройства сознания вплоть до развития комы и спустя 3 недели от момента госпитализации больная скончалась. В посеве крови выявлен рост Proteus mirabilis. На аутопсии определялись обширные некрозы печеночной паренхимы с выраженным холестазом, но с заметно редуцированным по сравнению с первичной биопсией воспалительным инфильтратом.
Комментируя данный клинический случай, A. Schattner et al. указывают, что наиболее вероятной причиной развития острой печеночной недостаточности, ставшей для пациентки фатальной, явился нимесулид. Это утверждение основано на факте внезапного начала заболевания, а также на исключении вирусного гепатита, токсического воздействия других лекарственных препаратов и гепатотоксичных ядов в качестве возможных причин возникновения острой печеночной недостаточнеости. Небезынтересно, что возникновение гепато–ренального синдрома и острой почечной недостаточности авторами связывается с наличием у нимесулида нефротоксичности. A. Schattner et al. констатируют, что представленный клинический случай лишь дополняет печальный список отчетов о возникновении фульминантной печеночной недостаточности, связанной с приемом нимесулида в целом ряде государств, начатый еще в 1998 г. K. Rainsford [40]. Доказанная связь терапии нимесулидом с возникновением острой печеночной недостаточности послужила поводом к принятию декларативных запретов на применение препарата в Японии, Индии, Шри–Ланке, Израиле, Финляндии, Испании и недопущению препарата к регистрации в США, Канаде, Великобритании, Австралии и Новой Зеландии. Так, например, в Финляндии частота индуцированных нимесулидом серьезных побочных эффектов (гепато– и нефротоксичность) составила 100 на 100000 населения в год, тогда как частота серьезных побочных эффектов при использовании других НПВП не превышает 1–27 на 100000 населения в год [6,33,40,43,45,46,50].
Говоря о терапии больных с НПВП–индуцированным повреждением печени, следует признать, что средств и методов, принципиально влияющих на исходы лечения, в настоящее время нет. Лечение проводится по стандартной схеме и заключается в отмене НПВП, создании пациенту охранительного режима с соблюдением диеты и ограничением физической нагрузки, назначении гепатопротекторов, витаминов, антиоксидантов и в наблюдении за динамикой печеночных функциональных тестов. При развитии фульминантной печеночной недостаточности и гепаторенального синдрома летальность достигает 80%, несмотря на проведение интенсивной посиндромной терапии, включая эктракорпоральную детоксикацию. Глюкокортикоиды могут быть полезны при явной аллергической природе гепатотоксичности, но их эффективность не доказана в контролируемых исследованиях. Теоретически они могут быть использованы у пациентов с позитивным антинуклеарным фактором или при вирусной этиологии предшествовавшего НПВП–повреждению хронического диффузного заболевания печени [1,2,6,38,47]. При тяжелом холестазе, обусловленном приемом НПВП, может быть эффективным назначение урсодезоксихолевой кислоты в сочетании с дюфалаком [34,47]. В любом случае клинически значимого НПВП–индуцированного повреждения печени необходим контроль и коррекция системы гемокоагуляции. Практически все зарубежные авторы возникновение острой печеночной недостаточности вследствие терапии НПВП считают прямым показанием к неотложной трансплантации печени как единственно эффективному лечебному мероприятию, способному сохранить больному жизнь.
Возможность проведения мониторинга специфических маркеров повреждения печени во время терапии НПВП также представляется трудноосуществимой: неясно у каких пациентов конкретно, как часто и за какой период следует осуществлять контроль. Справедливости ради следует заметить, что группы риска, где проявление гепатотоксического потенциала НПВП наиболее вероятно, предположительно определены. По данным L. Garcia–Rodriguez et al. (1994), C. Sgro et al. (2002), I. Lacroix et al. (2004), к факторам риска развития НПВП–индуцированного повреждения печени следует относить [18,21,46]: женский пол пациента, возраст старше 56 лет, наличие хронического аутоиммунного заболевания, наличие хронического диффузного заболевания печени, снижение функции почек, гипоальбуминемию, терапию высокими дозами НПВП, наличие хронического заболевания, требующего приема НПВП, полипрагмазию. С другой стороны, быть может, с клинической, да и с экономическойточки зрения целесообразнее не пассивно контролировать возможные проявления гепатотоксичности (которая очень быстро сама по себе способна стать неконтролируемым процессом), а исключить саму причину гепатотоксичности?! Тем более, что для современной клинической практики доступен целый ряд НПВП, чья гепатотоксичность выражена в минимальной степени.
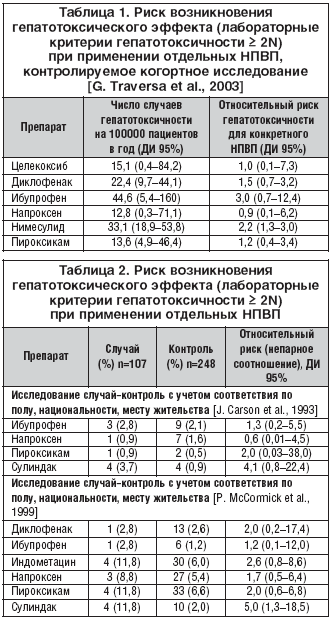
В обширном аналитическом отчете J. Rubenstein et al. (2004), являющемся результатом мета–анализа 8 клинических исследований гепатотоксичности НПВП, помимо установленного общего риска клинически значимого НПВП–индуцированного повреждения печени в популяции (4,8–8,6/100000 пациентов в год), приводятся данные об индивидуальных различиях гепатотоксического потенциала современных НПВП (табл. 1, 2) [43,50].
Из представленных данных следует, что наибольшим потенциалом в плане НПВП–индуцированной гепатотоксичности обладают сулиндак и индометацин. Наименьшей гепатотоксичностью, исходя из результатов проведенных исследований, обладают селективный ингибитор ЦОГ–2 целекоксиб и традиционный НПВП напроксен. Подтверждением того, что целекоксиб является одним из наименее гепатоксичных НПВП, являются результаты мета–анализа 14 многоцентровых исследований, проведенных в Европе и Северной Америке и посвященных оценке побочных эффектов НПВП у больных с остеоартритом и ревматоидным артритом. При этом частота и выраженность гепатотоксических эффектов целекоксиба (400 мг/сут.) сопоставлялась с таковыми у напроксена (1000 мг/сут.), диклофенака (150 мг/сут.), ибупрофена (800 мг/сут.) или плацебо в течение курса терапии продолжительностью от 2 до 12 недель (табл. 3) [24,43,50].
Результаты мета–анализа свидетельствуют о том, что частота нежелательных побочных эффектов со стороны печени (в том числе и серьезных) при терапии целекоксибом не отличается от таковой при применении плацебо и достоверно ниже, чем при терапии традиционными НПВП. Аналогичные результаты были получены при сопоставлении частоты нежелательных побочных эффектов целекоксиба, диклофенака и ибупрофена в известном многоцентровом исследовании CLASS (Celecoxib Long–term Arthritis Safety Study), охватывавшем более 8000 пациентов с остеоартритом и ревматоидным артритом. Частота нежелательных побочных эффектов со стороны печени при терапии диклофенаком оказалась в 4,7–5,3 раза выше, чем при терапии целекоксибом или ибупрофеном. Различие частоты нежелательных побочных эффектов со стороны печени при терапии целекоксибом и ибупрфеном оказалось недостоверным [24,50].
Согласно современным представлениям о синтропиях, анатомо–физиологическое единство гепато–билиарной системы и желудочно–кишечного тракта обусловливает развитие заболеваний в ЖКТ при наличии патологического процесса в печени и наоборот. Исследованиями второй половины двадцатого века было показано, что среди причин развития изъязвлений слизистой оболочки гастродуоденальной зоны заболевания печени – один из самых значимых факторов наряду с хроническими неспецифическими заболеваниями легких и заболеваниями сердечно–сосудистой системы [1,3,7,8,12]. При скрининговом проведении эндоскопического исследования у больных хроническими гепатитами и циррозом печени было установлено наличие острых или хронических язв в 52,2–75,0% случаев, тогда как варикозное расширение вен пищевода и желудка отмечалось только у 40,0% пациентов [12,19,32]. Формирование диффузных воспалительно–дистрофических заболеваний печени способствует развитию патологических изменений слизистой гастродуоденальной зоны задолго до манифестации гемоциркуляторных нарушений. При этом развитие изъязвлений слизистой одинаково выражено у мужчин и женщин и корелирует по частоте со степенью декомпенсации функций печени в градациях по Child–Pugh [8,32].
Г.В. Супорик и С.Г. Кочетков (2007), проведя целенаправленное эндоскопическое обследование 125 пациентов с хроническими диффузными заболеваниями печени, установили, что все больные имели сочетанную гастро–дуоденальную патологию. Практически у всех обследованных пациентов выявлено эрозивное поражение желудка, локализованное преимущественно в антральном отделе. Частота острых язв достигала 10,5%, при этом язвы были локализованы только в желудке. Микроскопически в биоптатах слизистой оболочки были обнаружены явления дистрофии поверхностного и ямочного эпителия, атрофические изменения фундальных и антральных желез, участки некроза в слизисто–подслизистом слое [7]. По данным F. Di Mario et al. (1998), у больных с хроническими диффузными заболеваниями печени острые язвы желудка выявлены в 40,5% случаев, острые язвы двенадцатиперстной кишки – в 48,6%, сочетанные желудочные и дуоденальные язвы – в 10,9%. Автор подчеркивает, что эрозии и изъязвления слизистой оболочки гастродуоденальной зоны встречаются почти у всех больных с портальной гипертензивной гастропатией [8].
Портальная гастропатия (портальная гипертензионная гастропатия, ПГГ) – термин, отражающий состояние сосудов подслизистого слоя и собственно слизистой оболочки гастродуоденальной зоны в условиях портальной гипертензии (рис. 2).
При ПГГ формируются множественные анастомозы между микрососудистым руслом слизистой оболочки желудка, расширенными венами, прекапиллярами желудка и пищевода. В условиях портальной гипертензии кровоток в стенке желудка усиливается, однако при этом слизистая желудка находится в состоянии хронической ишемии, что ослабляет защитный потенциал слизистой оболочки и предрасполагает к ее повреждениям. В случае ПГГ использование термина «гастропатия», по мнению большинства авторов, предпочтительнее использования термина «гастрит», поскольку основным морфологическим признаком ПГГ является эпителиальное и эндотелиальное повреждение без воспалительных изменений [1,7,8,32,47]. J. Krigeis (2005) указывает, что кровотечения из эрозий слизистой при ПГГ могут составлять до 23% от всех кровотечений при циррозах печени [12]. Отмечено, что частота ПГГ повышается при прогрессировании диффузных заболеваний печени и тесно коррелирует с наличием пищеводных и желудочных варикозов. Желудочно–кишечные кровотечения, обусловленные портальной гипертензией, как причина смерти больных циррозом печени стоят на втором месте после кровотечений, обусловленных варикозом вен пищевода и желудка. Кровотечения при портальной гастропатии ведут к развитию анемии, что нарушает функцию печени, а с другой стороны, являются фактором, ведущим к развитию печеночной энцефалопатии [3].
Рассматривая механизмы повреждения слизистой оболочки гастродуоденальной зоны при заболеваниях печени, большинство авторов используют концепцию «весов Shay», объясняющую ульцерогенез с позиции нарушения равновесия между факторами агрессии и факторами защиты. Очевидно, что агрессивность кислотно–пептического фактора при заболеваниях печени не изменяется, однако заметно снижается защитный потенциал слизистой оболочки гастродуоденальной зоны. Последнее обстоятельство является следствием нарушений гемоперфузии слизисто–подслизистого слоя как при острых, так и при хронических диффузных поражениях печени. В первом случае возникновение острой печеночной недостаточности приводит к редукции висцерального кровотока и возникновению типичного ишемического повреждения гастродуоденальной слизистой с развитием острых эрозий и язв. В случае хронических диффузных заболеваний печени развитие застойных явлений в системе портальных сосудов значимо меняет объемную скорость кровотока в микроциркуляторном русле всех слоев стенки полых органов пищеварительного тракта, особенно в зоне локализации вен–анастомозов. Нарушения микроциркуляции приводят к развитию циркуляторной гипоксии с нарушением транспорта кислорода и энергетических субстратов, отеку паравазального интерстиция. Вследствие этого существенно снижается регенераторный потенциал слизистой оболочки, что дает возможность реализации повреждающего действия кислотно–пептического фактора и ведет к развитию эрозивно–язвенных поражений в гастродуоденальной зоне.
В основе НПВП–индуцированного повреждения слизистой оболочки пищеварительной трубки находится угнетение традиционными нестероидными препаратами синтеза простагландинов Е2 и I2 (PgЕ2, PgI2) из арахидоновой кислоты путем ингибирования изофермента циклооксигеназы 1 типа (ЦОГ–1). К эффектам PgЕ1 относятся продукция адекватной по количеству и качеству слизи, секреция в просвет желудка бикарбонатов, сохранение достаточного объемного кровотока в слизисто–подслизистом слое, обеспечение репарации слизистой путем поддержания активной пролиферации камбиальных элементов желудочного эпителия. Из этого следует, что закономерным следствием терапии традиционными НПВП является снижение защитного потенциала слизистой пищеварительного тракта, что и приводит к развитию эрозивно–язвенного поражения. Согласно концепции «весов Shay», в данном случае к повреждению гастродуоденальной слизистой на фоне нормо– или гипоацидного состояния приводит именно снижение защитного потенциала слизистой. Кислотно–пептическое воздействие при формировании НПВП–индуцированных эрозий и язв играет роль производящего фактора [1,6].
Интересен тот факт, что возникающая при обусловленной токсическим действием НПВП острой печеночной недостаточности вазоконстрикция с гипоперфузией висцерального и почечного бассейнов некоторыми авторами также связывается с нарушением синтеза простагландинов под действием тех же самых НПВП [10,45,47]. В условиях снижающегося при острой печеночной недостаточности эффективного объема циркулирующей крови особенно важными являются регионарные вазодилатирующие эффекты простагландинов, синтезируемых при участии конститутивной ЦОГ–1. Угнетение синтеза простагландинов неселективными НПВП приводит к дисбалансу в системе эндотелины (вазоконстрикция) – простагландины (вазодилатация) с возникновением длительной ишемии. Закономерным следствием острых нарушений регионарной гемоперфузии являются: развитие острого тубулярного некроза и почечной недостаточности, формирование очагов ишемического некроза в слизисто–подслизистом слое пищеварительной трубки с возникновением острых эрозий и язв.
Объединив основные звенья патогенеза НПВП–индуцированного повреждения печени, сопровождающегося развитием острой или усугублением хронической печеночной недостаточности, синдрома портальной гипертензии, имеющие своим следствием повреждение слизистой оболочки пищеварительного тракта, а также непосредственный гастротоксический потенциал НПВП, не трудно представить возможную последовательность событий, приводящих в конечном итоге к формированию острого эрозивно–язвенного повреждения и развитию его осложнений: редукция висцерального артериального кровотока или хроническая гипертензия в системе воротной вены, нарушение гемоперфузии в стенке желудка и ДПК, ишемия и снижение защитного потенциала слизистой оболочки, формирование эрозий и язв под действием кислотно–пептического фактора, развитие кровотечения в условиях дефицита факторов свертывания и гипокоагуляции (рис. 3, 4).
В заключение считаем необходимым еще раз подчеркнуть, что гепатотоксичность и гастротоксичность НПВП являются патогенетически взаимосвязанными процессами. В отличие от токсического действия НПВП на слизистую пищеварительной трубки, механизм которого имеет четкое и логичное объяснение, рассуждения о причинах гепатотоксического эффекта НПВП в настоящее время весьма ограниченны нашими скромными представлениями о метаболических процессах в гепатоцитах. Тем не менее абсолютно недопустимо объяснять нежелательные эффекты со стороны печени при терапии НПВП реакциями «идиосинкразии», когда гепатотоксичность представляется как некое исключительное и непредсказуемое явление. Поэтому при внезапном возникновении безболевой желтухи, помимо серологических тестов и ультразвукового исследования, нелишним будет уточнить анамнез пациента и исключить лекарственное поражение печени, поскольку многие пациенты, благодаря широкой рекламе, относятся к некоторым НПВП как к пищевым добавкам и не считают необходимым поставить лечащего врача в известность об употреблениинестероидных препаратов. В случае, если НПВП–индуцированное повреждение печени является наиболее вероятной причиной печеночной недостаточности, помимо ряда стандартных в данной ситуации мероприятий, нелишним будет удостовериться и в интактности слизистой оболочки гастродуоденальной зоны. Тем не менее, учитывая крайне ограниченную возможность эффективного лечения пациентов с острой печеночной недостаточностью, единственным вариантом решения проблемы НПВП–индуцированной гепатотоксичности (равно как и в случае НПВП–индуцированной гастропатии) принципиально может являться только целенаправленная профилактика ее возникновения, основанная на применении НПВП с минимальным токсическим потенциалом.



Литература
1. Болезни органов пищеварения. В кн.: Руководство по медицине (The Merck Manual), под ред. Беркоу Р, Флетчер Э.Дж. М., 1997, с. 503–587.
2. Клиническая фармакология. В кн.: Руководство по медицине (The Merck Manual), под ред. Беркоу Р, Флетчер Э.Дж. М., 1997, с. 733–789.
3. Краснова М. В, Локтионова С. М, Пузакова О. Ю. Морфологические изменения слизистой желудка у пациентов с циррозами печени. Медицина в Кузбассе. – 2005. – Спецвып №2. – С. 89–90.
4. Муравьев Ю.В. Гепатоксичны ли нестероидные противовоспалительные препараты? Науч.–практ. ревмат. 2002, № 4, с.36 – 41.
5. Насонова В.А., Эрдес Ш.О. О всемирной декаде костно–суставных заболеваний 2000 – 2010. Научн.–практ. Ревматол. 2000, №4, с. 14–16.
6. Свиницкий А.С., Хомченкова Н.И., Пузанова О.Г. Ингибиторы ЦОГ–2: панацея от побочных эффектов нестероидных противовоспалительных препаратов или новые проблемы? Сучасна гастроэнтерологiя. 2003, №1 (11), с. 11–15.
7. Супорник Г.В., Кочетков С.Г. Особенности гастродуоденальной патологии у больных с хроническими диффузными заболеваниями печени. Вестник СамГУ, 2007, № 2 (52), с. 277 – 284.
8. Ханевич М.Д., Хрупкин В.И., Жерлов Г.К. Кровотечения из хронических гастродуоденальных язв у больных с внутрипеченочной портальной гипертензией. Новосибирск, 2003, 198 с.
9. Aithal PG, Day CP. The natural history of histologically proved drug induced liver disease. Gut 1999; 44:731–5.
10. Banks AT, Zimmerman HJ, Ishak KG, et al. Diclofenac–associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 1995; 22:820–7.
11. Bareille MP, Montastruc JL, Lapeyre–Mestre M. Liver damage and nonsteroidal anti–inflammatory drugs: case non–case study on the French pharmacovigilance database. [in French] Therpaie 2001; 56:51–5.
12. Bhala N., Plevris JN. Portal hypertensive enteropathy; an usual cause of gastrointestinal bleeding in cirrhosis: or is it? JR Coll Physicians, 2006; 36; 208 – 210.
13. Bjorkman D. Nonsteroidal anti–inflammatory drug associated toxicity of the liver, lower gastrointestinal tract and the esophagus. Am J Med 1998; 105:17–21S.
14. Bort R, Ponsoda X, Jover R, et al. Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther 1999; 288:65–72.
15. Danan G, Benichou Ch. Causality assessment of adverse reactions to drugs – I. A novel method based on the conclusions of international consensus meetings: applications to drug–induced liver injury. J. Clin Epidemiol, 1993; 4: 111–4.
16. Dierkes–Globisch A, Schafer R, Mohr HH. Asymptomatic diclofenac–induced acute hepatitis [in German]. Dtsch Med Wochenschr 2000; 125:797–800.
17. Fontana RJ, McCashland TM, Benner KG, et al. Acute liver failure associated with prolonged use of bromfenac leading to liver transplantation. The Acute Liver Failure Study Group. Liver Transpl Surg 1999; 5:480–4.
18. Garcia Rodriguez LA, Perez Guttan. The role of non–steroidal anti–inflammatory drugs in acute liver injury. BMJ, 1994; 6: 107–17.
19. Gastritis and Other Gastropathies. In: Feldman, M.; Friedman, L.S.; Sleisenger, M.H. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology/Diagnosis/Management. 7th ed. [2–volume set]. St. Louis, MO: Saunders. 2002. p. 810–827.
20. Greaves RR, Agarwal A, Patch D, et al. Inadvertent diclofenac rechallenge from generic and non–generic prescribing, leading to liver transplantation for fulminant liver failure. Eur J Gastroenterol Hepatol 2001; 13:71–3.
21. Lacroix I, Lapeyre–Mestre M, Bagheri H. Nonsteroidal anti–inflammatory drug–induced liver injury: a case–control study in primary care. Fundamental & Clinical Pharmacology, 2004; 18: 201–6.
22. Lapeyre–Mestre M, Rueda de Castro AM. Non–steroidal anti–inflammatory drug–related hepatic damage in France and Spain. Fundamental & Clinical Pharmacology, 2006; 20: 391–5.
23. Mabee CL, Mabee SW, Baker PB. Fulminant hepatic failure associated with etodolac use. Am J Gastroenterol, 1995; 90: 659–61.
24. Maddrey WC, Maurath CJ, Verburg KM, et al. The hepatic safety and tolerability of the novel cyclo–oxygenase–2 inhibitor celecoxib. Am J Ther 2000; 7:153–8.
25. Martin R, Biswas P. The incidence of adverse events and risk factors for upper gastriointestinal disorders associated with meloxicam. Br J Clin Pharmacol, 2000; 50: 35–42.
26. Masubuchi Y, Horie T. A possible mechanisms of naproxen–induced ipid–peroxidation in liver microsomes. Pharmacol Toxicol, 2001; 89: 43–8.
27. Masubuchi Y, Horie T. Structural requirements for the hepatotoxicity of nonsteroidal anti–inflammatory drugs. J Pharmacol Exp Ther, 1998: 287: 208–13.
28. Masubuchi Y, Nakayama S, Horie T. Roe of mitochondrial permeability transition in diclofenac–induced hepatotoxicity. Hepatology, 2002; 35: 544–51.
29. Masubuchi Y, Nakayama S, Horie T. Role of mitochondrial permeability transition in diclofenac–induced hepatotoxicity in rats. Hepatology 2002; 35:544–551.
30. Masubuchi Y, Yamada S, Horie T. Possible mechanisms of hepatocyte injury induced by diphenylamine and its structurally related NSAIDs. J Pharmacol Exp Ther, 2000; 292: 982–7.
31. Mc Cormick PA, Kennedy F, Curry M. COX 2 inhibitor and fulminant hepatic failure. Lancet, 1999; 353: 40–1.
32. McCormack TT, Sims J, Eyre–Brook I, et al. Lesions in portal hypertension: inflammatory gastritis or congestive gastropathy? Gut 1985;26:1226–32.
33. Merlani G, Fox M, Oehen HP, Cathomas G, Renner EL, Fattinger K, Schneeman M, Kullak–Ublick GA. Fatal hepatotoxicity secondary to nimesulide. Eur J Clin Pharmacol 2001; 57:321–6.
34. O’Connor N, Dargan PI, Jones AL. Hepatocellular damage from non–steroidal anti–inflammatory drugs. Q J Med, 2003: 96: 787–791.
35. Ponsoda X, Pareja E, Gomez–Lechon MJ, et al. Drug biotransformation by human hepatocytes. In vitro/in vivo metabolism by cells from the same donor. J Hepatol 2001; 34:19–24.
36. Purdum PP 3rd, Shelden SL, Boyd JW, Shiffman ML. Oxaprozin–induced fulminant hepatitis. Ann Pharmacother 1994; 28:1159–61.
37. Rabinovitz M, Van Thiel DH. Hepatotoxicity of non–steroidal anti–inflammatory drugs. Am J Gastroenterol 1992; 87: 1696–704.
38. Rabkin JM, Smith MJ, Orloff SL, et al. Fatal fulminant hepatitis associated with bromofenac use. Ann Pharmacother 1999; 33:945–7.
39. Rabkin JM, Smith MJ, Orloff SL. Fatal fulminant hepatitis associated with bromfenac use. Ann Pharmacother, 1999; 33: 945–7.
40. Rainsford KD. An analysis from clinico–epidemiological data of the principal adverse events from the COX–2 selective NSAID, nimesuide, with particular reference of hepatic injury. Inflammopharmacology, 1998; 6: 203–21.
41. Riley TR, Smith JP. Ibuprofen–induced hepatotoxicity in patients with chronic hepatitis C: A case series. Am J Gastroenterol 1998; 93:1563–5.
42. Rodriguez–Gonzalez FJ, Montero JL, Puente J, et al. Orthotopic liver transplantation after subacute liver failure induced by therapeutic doses of ibuprofen. Am J Gastroenterol 2002; 97:2476–7.
43. Rubenstein J., Laine L. Systematic review: the hepatotoxicity of non–steroidal anti–inflammatory drugs. Aliment Pharmacol Ther, 2004; 20: 373–80.
44. Samonakis DN, Triantos CK, Thalheimer U: Management of portal hypertension. Postgrad Med J 2004 Nov; 80(949): 634–41.
45. Schattner A, Sokolovskaya N. 2000
46. Sgro C, Clinard F, Quazir K. Incidence of drug–induced hepatic injuries. Hepatology, 2002; 36: 451–5.
47. Sherlock S, Dooley J, eds: Diseases of the Liver and Biliary System. Oxford, UK: Blackwell Science; 1997.
48. Tan H, Ong WM, Lai SH. Nimesulide–induced hepatotoxicity and fatal hepatic failure. Singapore Med J, 2007; 48(6): 582–5.
49. Teoh NC, Farrell GC. Hepatotoxicity associated with non–steroidal anti–inflammatory drugs. Clin Liver Dis, 2003; 7: 401–13.
50. Traversa G, Bianchi C, DaCas R. Cohort study of hepatotoxicity associated with nimesulide and other non–steroidal anti–inflammatory drugs. BMJ, 2003; 327: 18–22.
51. Wongcharatrawee S, Groszmann RJ: Diagnosing portal hypertension. Baillieres Best Pract Res Clin Gastroenterol 2000 Dec; 14(6): 881–94.
Ключевыми моментами данной патогенетической модели являются: прямое токсическое влияние НПВП на интактную или патологически измененную печеночную паренхиму с возникновением острой или усугублением хронической печеночной недостаточности, портальная гастропатия у больных с циррозом печени и синдромом портальной гипертензии, НПВП–индуцированное повреждение слизистой гастродуоденальной зоны.
Прямое токсическое действие НПВП на печень. Лекарственное поражение печени, в том числе и НПВП, является одной из серьезных проблем современной гепатологии. Это обусловлено не столько частотой возникновения нежелательных реакций, сколько большой вероятностью неблагоприятных исходов в случае их возникновения: до 25% всех случаев фульминантной печеночной недостаточности связано именно с лекарственным поражением печени. НПВП, являясь с химической точки зрения крайне неоднородной группой лекарственных препаратов, оказывают сходное терапевтическое действие и вызывают однотипные побочные эффекты. N. O’Connor et al. (2003) на основании изучения эпидемиологических аспектов гепатотоксичности нестероидных препаратов сделали вывод о том, что почти все современные НПВП качественно обладают гепатотоксичностью, количественно выраженной в большей или меньшей степени. Несмотря на то что относительный риск клинически значимого повреждения печени вследствие применения НПВП относительно невысок (8–27 случаев на 100000 пациентов в год), последствия возникшего НПВП–индуцированного повреждения печени зачастую становятся самыми серьезными: фульминантная печеночная недостаточность и гепаторенальный синдром [35]. Указанные нежелательные побочные эффекты терапии НПВП, следствием которых в конечном итоге явились гибель пациентов или необходимость экстренных трансплантаций печени, послужили причиной запрещения на использование в клинической практике беноксапрофена, дроксикама, бромфенака, пирпрофена, фенклофенака и в ряде государств нимесулида [10,11,15,17,21–23,31,33,36,38–40,42,45].
Считается, что повреждение печеночной паренхимы вследствие приема НПВП может возникать в различный временной промежуток: непосредственно после начала терапии, в процессе двух–трехнедельного курса лечения, спустя недели или даже месяцы после его завершения. P. Aithal et al. (1999) полагают, что наиболее часто клинико–лабораторные проявления НПВП–индуцированного повреждения печени находятся в интервале 6–12 недель с момента начала терапии. I. Lacroix et al. (2004), анализируя особенности клинической манифестации острых повреждений печени, связанных с приемом НПВП, отмечает, что у 5–27% пациентов такие повреждения протекают бессимптомно и могут быть выявлены лишь по повышению активности трансаминаз плазмы [9,21]. Большинство исследователей подчеркивает, что риск лекарственного поражения печени возрастает при наличии хронического диффузного заболевания печени любой этиологии. При этом нарушение метаболизма лекарств прямо пропорционально выраженности хронической печеночноклеточной недостаточности [4,22,35,41,48].
Как указывают H. Tan et al. (2007), клинико–морфологические формы токсического воздействия НПВП на печень весьма вариабельны: острый гепатит, холестаз, холестатический гепатит, имеющий тенденцию к хронизации гранулематозный гепатит, хронический холестаз с дуктопенией, желтая атрофия печени, проявляющаяся фульминантной печеночной недостаточностью. Авторы полагают, что наиболее характерной особенностью морфологической картины при НПВП–индуцированном повреждении печени является сочетание обширного гепатоцеллюлярного некроза с перипортальной мононуклеарной инфильтрацией и холестатическим компонентом (рис. 1) [48].
Несмотря на немалое количество исследований, посвященных проблеме НПВП–индуцированной гепатотоксичности, собственно механизм повреждения печеночной паренхимы на клеточном и субклеточном уровнях сегодня остается предметом дискуссии [5].
N. O’Connor et al. (2003) приводит две наиболее часто упоминающиеся в литературе причины гепатотоксичности НПВП: 1) реакции гиперчувствительности, 2) метаболическое повреждение (аберрация). О возможности НПВП–индуцированного повреждения печени, являющегося следствием реакций гиперчувствительности (аллергических реакций), по мнению R. Greaves et al. (2001), свидетельствует факт развития «перекрестной» токсичности при разделенных во времени курсах терапии химически сходными препаратами (диклофенак – тиапрофеновая кислота), а также факт клинического проявления гепатотоксичности у препарата при повторном курсе терапии [20,34]. R. Bort et al. (1999) на основании обобщения экспериментальных данных представили последовательность событий при повреждении гепатоцита НПВП следующим образом. Основной мишенью НПВП на субклеточном уровне являются митохондрии. В процессе метаболизма НПВП цитохромом Р450 образуются производные НПВП (например, 5–гидроксидиклофенак и n,5–дигидроксидиклофенак), которые способны влиять на процессы переноса электронов в дыхательной цепи на кристах митохондрий, приводя к нарушению окислительного фосфорилирования, синтеза АТФ и энергетическому дефициту в клетке. Не исключено, что токсическим влиянием на митохондрии обладают и нативные НПВП. Нарушение процессов окислительного фосфорилирования в митохондриях и микросомальное окисление некоторых НПВП (например, напроксена) приводят к активации свободно–радикального окисления, перекисному окислению липидов мембран, НАДФ и тиоловых групп белков, результатом чего, в конечном счете, является дезорганизация мембран, гибель гепатоцита и синтопичных клеточных структур (клетки желчных протоков) [14,26–30]. Возможно, что в процессе дезорганизации цитолемма приобретает антигенные свойства, что приводит к индукции аутоиммунного ответа и морфологически проявляется как перипортальный отек и момонуклеарная инфильтрация.
Типичным примером гепатотоксического действия НПВП может служить представленный A. Schattner et al. (2000) случай развития острой печеночной недостаточности и гепаторенального синдрома, этиологически связанный с приемом нимесулида [45].
70–летняя пациентка была госпитализирована по поводу выраженной слабости и клинически манифестированной желтухи, развившихся за 2 суток до госпитализации. Из объективных симптомов в момент госпитализации были выявлены желтушность кожи и иктеричность склер, тахикардия (117 в мин.). Больная страдала ожирением и остеоартритом коленных суставов, по поводу которого в течение нескольких лет принимала диклофенак. Впоследствии диклофенак был заменен нимесулидом (100 мг дважды в сутки), 5–дневный курс терапии которым был завершен за две недели до настоящей госпитализации. Лабораторные показатели на момент госпитализации: гемоглобин 135 г/л, лейкоциты 16х109/л (лейкоцитарная формула не изменена), тромбоциты 203х109/л, СОЭ 28 мм/ч, азот мочевины 5,8 ммоль/л, креатинин плазмы 70,7 мкмоль/л, электролиты плазмы и биохимический анализ мочи (за исключением желчных пигментов) в пределах нормы; общий билирубин плазмы 216 мкмоль/л, алкалин фосфатаза 285 МЕ/л, ?–ГТП 173 МЕ/л, АсАТ 1700 МЕ/л, АлАТ 1240 МЕ/л, ЛДГ 1460 МЕ/л, ?–амилаза сыворотки 285 МЕ/л, общий холестерол 3,9 ммоль/л, триглицериды 1,6 ммоль/л; альбумин сыворотки 25 г/л, глобулины 40 г/л; протромбиновое время 21с, МНО 2,3, АЧТВ 33 с (впоследствии не изменялось при внутривенном введении витамина К). Серологические пробы на вирусные гепатиты А, В, С и Е, вирус простого герпеса, ВИЧ, ПЦР на ДНК HBV и РНК HCV отрицательные. Инфекционная этиология заболевания была исключена, аутоантитела не выявлены, скрининг–тесты на экзогенные токсические агенты отрицательные. Биохимический анализ крови, проведенный за месяц до госпитализации, отклонений от нормы не выявил. Выполненные непосредственно после госпитализации ультразвуковое исследование и компьютерная томография брюшной полости отклонений от нормы не выявили. При гистологическом исследовании ткани печени, полученной при пункционной тонкоигольной биопсии на 5–й день нахождения в стационаре, были выявлены массивные некрозы гепатоцитов с выраженной смешанной (плазматические клетки, лимфоциты, эозинофилы и гранулоциты) воспалительной инфильтрацией портальных трактов и паренхимы. Данная картина согласовывалась с клиническими признаками фульминантного гепатита. К концу первой недели пребывания в стационаре отмечено нарастание желтухи с повышением общего билирубина до 484 мкмоль/л (максимальный общий билирубин 564 мкмоль/л) при снижении уровня печеночных ферментов более чем наполовину от их первоначальных значений, отмечено внезапное развитие гипогликемии. На четвертый день нахождения в стационаре у пациентки выявлено повышение азота мочевины до 10,8 ммоль/л и креатинина сыворотки до 124 мкмоль/л с последующим нарастанием к концу первой недели до 177 мкмоль/л. Это сопровождалось возникновением олигурии (300 мл/сут.) при неизмененном осадке мочи, FENa 0,1%, отсутствием ответа на водную нагрузку. Проведение терапии глюкокортикоидами (метилпреднизолон внутривенно 125 мг/сут.), назначение урсодезоксихолевой кислоты, лактулозы, диета с низким содержанием белка, впоследствии – инфузии глюкозы и допамина видимого эффекта не оказали. Прогрессировали гипоальбуминемия, коагулопатия, нарушения ментального статуса. Дважды отмечалась мелена. При дальнейшем нарастании почечной недостаточности (повышение креатинина плазмы до 495 мкмоль/л, лейкоцитурия, микрогематурия, FENa 2%) дважды проводились сеансы гемодиализа. Произведен посев крови на стерильность, назначены антибиотики широкого спектра действия. Однако состояние пациентки ухудшалось, нарастали расстройства сознания вплоть до развития комы и спустя 3 недели от момента госпитализации больная скончалась. В посеве крови выявлен рост Proteus mirabilis. На аутопсии определялись обширные некрозы печеночной паренхимы с выраженным холестазом, но с заметно редуцированным по сравнению с первичной биопсией воспалительным инфильтратом.
Комментируя данный клинический случай, A. Schattner et al. указывают, что наиболее вероятной причиной развития острой печеночной недостаточности, ставшей для пациентки фатальной, явился нимесулид. Это утверждение основано на факте внезапного начала заболевания, а также на исключении вирусного гепатита, токсического воздействия других лекарственных препаратов и гепатотоксичных ядов в качестве возможных причин возникновения острой печеночной недостаточнеости. Небезынтересно, что возникновение гепато–ренального синдрома и острой почечной недостаточности авторами связывается с наличием у нимесулида нефротоксичности. A. Schattner et al. констатируют, что представленный клинический случай лишь дополняет печальный список отчетов о возникновении фульминантной печеночной недостаточности, связанной с приемом нимесулида в целом ряде государств, начатый еще в 1998 г. K. Rainsford [40]. Доказанная связь терапии нимесулидом с возникновением острой печеночной недостаточности послужила поводом к принятию декларативных запретов на применение препарата в Японии, Индии, Шри–Ланке, Израиле, Финляндии, Испании и недопущению препарата к регистрации в США, Канаде, Великобритании, Австралии и Новой Зеландии. Так, например, в Финляндии частота индуцированных нимесулидом серьезных побочных эффектов (гепато– и нефротоксичность) составила 100 на 100000 населения в год, тогда как частота серьезных побочных эффектов при использовании других НПВП не превышает 1–27 на 100000 населения в год [6,33,40,43,45,46,50].
Говоря о терапии больных с НПВП–индуцированным повреждением печени, следует признать, что средств и методов, принципиально влияющих на исходы лечения, в настоящее время нет. Лечение проводится по стандартной схеме и заключается в отмене НПВП, создании пациенту охранительного режима с соблюдением диеты и ограничением физической нагрузки, назначении гепатопротекторов, витаминов, антиоксидантов и в наблюдении за динамикой печеночных функциональных тестов. При развитии фульминантной печеночной недостаточности и гепаторенального синдрома летальность достигает 80%, несмотря на проведение интенсивной посиндромной терапии, включая эктракорпоральную детоксикацию. Глюкокортикоиды могут быть полезны при явной аллергической природе гепатотоксичности, но их эффективность не доказана в контролируемых исследованиях. Теоретически они могут быть использованы у пациентов с позитивным антинуклеарным фактором или при вирусной этиологии предшествовавшего НПВП–повреждению хронического диффузного заболевания печени [1,2,6,38,47]. При тяжелом холестазе, обусловленном приемом НПВП, может быть эффективным назначение урсодезоксихолевой кислоты в сочетании с дюфалаком [34,47]. В любом случае клинически значимого НПВП–индуцированного повреждения печени необходим контроль и коррекция системы гемокоагуляции. Практически все зарубежные авторы возникновение острой печеночной недостаточности вследствие терапии НПВП считают прямым показанием к неотложной трансплантации печени как единственно эффективному лечебному мероприятию, способному сохранить больному жизнь.
Возможность проведения мониторинга специфических маркеров повреждения печени во время терапии НПВП также представляется трудноосуществимой: неясно у каких пациентов конкретно, как часто и за какой период следует осуществлять контроль. Справедливости ради следует заметить, что группы риска, где проявление гепатотоксического потенциала НПВП наиболее вероятно, предположительно определены. По данным L. Garcia–Rodriguez et al. (1994), C. Sgro et al. (2002), I. Lacroix et al. (2004), к факторам риска развития НПВП–индуцированного повреждения печени следует относить [18,21,46]: женский пол пациента, возраст старше 56 лет, наличие хронического аутоиммунного заболевания, наличие хронического диффузного заболевания печени, снижение функции почек, гипоальбуминемию, терапию высокими дозами НПВП, наличие хронического заболевания, требующего приема НПВП, полипрагмазию. С другой стороны, быть может, с клинической, да и с экономическойточки зрения целесообразнее не пассивно контролировать возможные проявления гепатотоксичности (которая очень быстро сама по себе способна стать неконтролируемым процессом), а исключить саму причину гепатотоксичности?! Тем более, что для современной клинической практики доступен целый ряд НПВП, чья гепатотоксичность выражена в минимальной степени.
В обширном аналитическом отчете J. Rubenstein et al. (2004), являющемся результатом мета–анализа 8 клинических исследований гепатотоксичности НПВП, помимо установленного общего риска клинически значимого НПВП–индуцированного повреждения печени в популяции (4,8–8,6/100000 пациентов в год), приводятся данные об индивидуальных различиях гепатотоксического потенциала современных НПВП (табл. 1, 2) [43,50].
Из представленных данных следует, что наибольшим потенциалом в плане НПВП–индуцированной гепатотоксичности обладают сулиндак и индометацин. Наименьшей гепатотоксичностью, исходя из результатов проведенных исследований, обладают селективный ингибитор ЦОГ–2 целекоксиб и традиционный НПВП напроксен. Подтверждением того, что целекоксиб является одним из наименее гепатоксичных НПВП, являются результаты мета–анализа 14 многоцентровых исследований, проведенных в Европе и Северной Америке и посвященных оценке побочных эффектов НПВП у больных с остеоартритом и ревматоидным артритом. При этом частота и выраженность гепатотоксических эффектов целекоксиба (400 мг/сут.) сопоставлялась с таковыми у напроксена (1000 мг/сут.), диклофенака (150 мг/сут.), ибупрофена (800 мг/сут.) или плацебо в течение курса терапии продолжительностью от 2 до 12 недель (табл. 3) [24,43,50].
Результаты мета–анализа свидетельствуют о том, что частота нежелательных побочных эффектов со стороны печени (в том числе и серьезных) при терапии целекоксибом не отличается от таковой при применении плацебо и достоверно ниже, чем при терапии традиционными НПВП. Аналогичные результаты были получены при сопоставлении частоты нежелательных побочных эффектов целекоксиба, диклофенака и ибупрофена в известном многоцентровом исследовании CLASS (Celecoxib Long–term Arthritis Safety Study), охватывавшем более 8000 пациентов с остеоартритом и ревматоидным артритом. Частота нежелательных побочных эффектов со стороны печени при терапии диклофенаком оказалась в 4,7–5,3 раза выше, чем при терапии целекоксибом или ибупрофеном. Различие частоты нежелательных побочных эффектов со стороны печени при терапии целекоксибом и ибупрфеном оказалось недостоверным [24,50].
Согласно современным представлениям о синтропиях, анатомо–физиологическое единство гепато–билиарной системы и желудочно–кишечного тракта обусловливает развитие заболеваний в ЖКТ при наличии патологического процесса в печени и наоборот. Исследованиями второй половины двадцатого века было показано, что среди причин развития изъязвлений слизистой оболочки гастродуоденальной зоны заболевания печени – один из самых значимых факторов наряду с хроническими неспецифическими заболеваниями легких и заболеваниями сердечно–сосудистой системы [1,3,7,8,12]. При скрининговом проведении эндоскопического исследования у больных хроническими гепатитами и циррозом печени было установлено наличие острых или хронических язв в 52,2–75,0% случаев, тогда как варикозное расширение вен пищевода и желудка отмечалось только у 40,0% пациентов [12,19,32]. Формирование диффузных воспалительно–дистрофических заболеваний печени способствует развитию патологических изменений слизистой гастродуоденальной зоны задолго до манифестации гемоциркуляторных нарушений. При этом развитие изъязвлений слизистой одинаково выражено у мужчин и женщин и корелирует по частоте со степенью декомпенсации функций печени в градациях по Child–Pugh [8,32].
Г.В. Супорик и С.Г. Кочетков (2007), проведя целенаправленное эндоскопическое обследование 125 пациентов с хроническими диффузными заболеваниями печени, установили, что все больные имели сочетанную гастро–дуоденальную патологию. Практически у всех обследованных пациентов выявлено эрозивное поражение желудка, локализованное преимущественно в антральном отделе. Частота острых язв достигала 10,5%, при этом язвы были локализованы только в желудке. Микроскопически в биоптатах слизистой оболочки были обнаружены явления дистрофии поверхностного и ямочного эпителия, атрофические изменения фундальных и антральных желез, участки некроза в слизисто–подслизистом слое [7]. По данным F. Di Mario et al. (1998), у больных с хроническими диффузными заболеваниями печени острые язвы желудка выявлены в 40,5% случаев, острые язвы двенадцатиперстной кишки – в 48,6%, сочетанные желудочные и дуоденальные язвы – в 10,9%. Автор подчеркивает, что эрозии и изъязвления слизистой оболочки гастродуоденальной зоны встречаются почти у всех больных с портальной гипертензивной гастропатией [8].
Портальная гастропатия (портальная гипертензионная гастропатия, ПГГ) – термин, отражающий состояние сосудов подслизистого слоя и собственно слизистой оболочки гастродуоденальной зоны в условиях портальной гипертензии (рис. 2).
При ПГГ формируются множественные анастомозы между микрососудистым руслом слизистой оболочки желудка, расширенными венами, прекапиллярами желудка и пищевода. В условиях портальной гипертензии кровоток в стенке желудка усиливается, однако при этом слизистая желудка находится в состоянии хронической ишемии, что ослабляет защитный потенциал слизистой оболочки и предрасполагает к ее повреждениям. В случае ПГГ использование термина «гастропатия», по мнению большинства авторов, предпочтительнее использования термина «гастрит», поскольку основным морфологическим признаком ПГГ является эпителиальное и эндотелиальное повреждение без воспалительных изменений [1,7,8,32,47]. J. Krigeis (2005) указывает, что кровотечения из эрозий слизистой при ПГГ могут составлять до 23% от всех кровотечений при циррозах печени [12]. Отмечено, что частота ПГГ повышается при прогрессировании диффузных заболеваний печени и тесно коррелирует с наличием пищеводных и желудочных варикозов. Желудочно–кишечные кровотечения, обусловленные портальной гипертензией, как причина смерти больных циррозом печени стоят на втором месте после кровотечений, обусловленных варикозом вен пищевода и желудка. Кровотечения при портальной гастропатии ведут к развитию анемии, что нарушает функцию печени, а с другой стороны, являются фактором, ведущим к развитию печеночной энцефалопатии [3].
Рассматривая механизмы повреждения слизистой оболочки гастродуоденальной зоны при заболеваниях печени, большинство авторов используют концепцию «весов Shay», объясняющую ульцерогенез с позиции нарушения равновесия между факторами агрессии и факторами защиты. Очевидно, что агрессивность кислотно–пептического фактора при заболеваниях печени не изменяется, однако заметно снижается защитный потенциал слизистой оболочки гастродуоденальной зоны. Последнее обстоятельство является следствием нарушений гемоперфузии слизисто–подслизистого слоя как при острых, так и при хронических диффузных поражениях печени. В первом случае возникновение острой печеночной недостаточности приводит к редукции висцерального кровотока и возникновению типичного ишемического повреждения гастродуоденальной слизистой с развитием острых эрозий и язв. В случае хронических диффузных заболеваний печени развитие застойных явлений в системе портальных сосудов значимо меняет объемную скорость кровотока в микроциркуляторном русле всех слоев стенки полых органов пищеварительного тракта, особенно в зоне локализации вен–анастомозов. Нарушения микроциркуляции приводят к развитию циркуляторной гипоксии с нарушением транспорта кислорода и энергетических субстратов, отеку паравазального интерстиция. Вследствие этого существенно снижается регенераторный потенциал слизистой оболочки, что дает возможность реализации повреждающего действия кислотно–пептического фактора и ведет к развитию эрозивно–язвенных поражений в гастродуоденальной зоне.
В основе НПВП–индуцированного повреждения слизистой оболочки пищеварительной трубки находится угнетение традиционными нестероидными препаратами синтеза простагландинов Е2 и I2 (PgЕ2, PgI2) из арахидоновой кислоты путем ингибирования изофермента циклооксигеназы 1 типа (ЦОГ–1). К эффектам PgЕ1 относятся продукция адекватной по количеству и качеству слизи, секреция в просвет желудка бикарбонатов, сохранение достаточного объемного кровотока в слизисто–подслизистом слое, обеспечение репарации слизистой путем поддержания активной пролиферации камбиальных элементов желудочного эпителия. Из этого следует, что закономерным следствием терапии традиционными НПВП является снижение защитного потенциала слизистой пищеварительного тракта, что и приводит к развитию эрозивно–язвенного поражения. Согласно концепции «весов Shay», в данном случае к повреждению гастродуоденальной слизистой на фоне нормо– или гипоацидного состояния приводит именно снижение защитного потенциала слизистой. Кислотно–пептическое воздействие при формировании НПВП–индуцированных эрозий и язв играет роль производящего фактора [1,6].
Интересен тот факт, что возникающая при обусловленной токсическим действием НПВП острой печеночной недостаточности вазоконстрикция с гипоперфузией висцерального и почечного бассейнов некоторыми авторами также связывается с нарушением синтеза простагландинов под действием тех же самых НПВП [10,45,47]. В условиях снижающегося при острой печеночной недостаточности эффективного объема циркулирующей крови особенно важными являются регионарные вазодилатирующие эффекты простагландинов, синтезируемых при участии конститутивной ЦОГ–1. Угнетение синтеза простагландинов неселективными НПВП приводит к дисбалансу в системе эндотелины (вазоконстрикция) – простагландины (вазодилатация) с возникновением длительной ишемии. Закономерным следствием острых нарушений регионарной гемоперфузии являются: развитие острого тубулярного некроза и почечной недостаточности, формирование очагов ишемического некроза в слизисто–подслизистом слое пищеварительной трубки с возникновением острых эрозий и язв.
Объединив основные звенья патогенеза НПВП–индуцированного повреждения печени, сопровождающегося развитием острой или усугублением хронической печеночной недостаточности, синдрома портальной гипертензии, имеющие своим следствием повреждение слизистой оболочки пищеварительного тракта, а также непосредственный гастротоксический потенциал НПВП, не трудно представить возможную последовательность событий, приводящих в конечном итоге к формированию острого эрозивно–язвенного повреждения и развитию его осложнений: редукция висцерального артериального кровотока или хроническая гипертензия в системе воротной вены, нарушение гемоперфузии в стенке желудка и ДПК, ишемия и снижение защитного потенциала слизистой оболочки, формирование эрозий и язв под действием кислотно–пептического фактора, развитие кровотечения в условиях дефицита факторов свертывания и гипокоагуляции (рис. 3, 4).
В заключение считаем необходимым еще раз подчеркнуть, что гепатотоксичность и гастротоксичность НПВП являются патогенетически взаимосвязанными процессами. В отличие от токсического действия НПВП на слизистую пищеварительной трубки, механизм которого имеет четкое и логичное объяснение, рассуждения о причинах гепатотоксического эффекта НПВП в настоящее время весьма ограниченны нашими скромными представлениями о метаболических процессах в гепатоцитах. Тем не менее абсолютно недопустимо объяснять нежелательные эффекты со стороны печени при терапии НПВП реакциями «идиосинкразии», когда гепатотоксичность представляется как некое исключительное и непредсказуемое явление. Поэтому при внезапном возникновении безболевой желтухи, помимо серологических тестов и ультразвукового исследования, нелишним будет уточнить анамнез пациента и исключить лекарственное поражение печени, поскольку многие пациенты, благодаря широкой рекламе, относятся к некоторым НПВП как к пищевым добавкам и не считают необходимым поставить лечащего врача в известность об употреблениинестероидных препаратов. В случае, если НПВП–индуцированное повреждение печени является наиболее вероятной причиной печеночной недостаточности, помимо ряда стандартных в данной ситуации мероприятий, нелишним будет удостовериться и в интактности слизистой оболочки гастродуоденальной зоны. Тем не менее, учитывая крайне ограниченную возможность эффективного лечения пациентов с острой печеночной недостаточностью, единственным вариантом решения проблемы НПВП–индуцированной гепатотоксичности (равно как и в случае НПВП–индуцированной гастропатии) принципиально может являться только целенаправленная профилактика ее возникновения, основанная на применении НПВП с минимальным токсическим потенциалом.
Литература
1. Болезни органов пищеварения. В кн.: Руководство по медицине (The Merck Manual), под ред. Беркоу Р, Флетчер Э.Дж. М., 1997, с. 503–587.
2. Клиническая фармакология. В кн.: Руководство по медицине (The Merck Manual), под ред. Беркоу Р, Флетчер Э.Дж. М., 1997, с. 733–789.
3. Краснова М. В, Локтионова С. М, Пузакова О. Ю. Морфологические изменения слизистой желудка у пациентов с циррозами печени. Медицина в Кузбассе. – 2005. – Спецвып №2. – С. 89–90.
4. Муравьев Ю.В. Гепатоксичны ли нестероидные противовоспалительные препараты? Науч.–практ. ревмат. 2002, № 4, с.36 – 41.
5. Насонова В.А., Эрдес Ш.О. О всемирной декаде костно–суставных заболеваний 2000 – 2010. Научн.–практ. Ревматол. 2000, №4, с. 14–16.
6. Свиницкий А.С., Хомченкова Н.И., Пузанова О.Г. Ингибиторы ЦОГ–2: панацея от побочных эффектов нестероидных противовоспалительных препаратов или новые проблемы? Сучасна гастроэнтерологiя. 2003, №1 (11), с. 11–15.
7. Супорник Г.В., Кочетков С.Г. Особенности гастродуоденальной патологии у больных с хроническими диффузными заболеваниями печени. Вестник СамГУ, 2007, № 2 (52), с. 277 – 284.
8. Ханевич М.Д., Хрупкин В.И., Жерлов Г.К. Кровотечения из хронических гастродуоденальных язв у больных с внутрипеченочной портальной гипертензией. Новосибирск, 2003, 198 с.
9. Aithal PG, Day CP. The natural history of histologically proved drug induced liver disease. Gut 1999; 44:731–5.
10. Banks AT, Zimmerman HJ, Ishak KG, et al. Diclofenac–associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 1995; 22:820–7.
11. Bareille MP, Montastruc JL, Lapeyre–Mestre M. Liver damage and nonsteroidal anti–inflammatory drugs: case non–case study on the French pharmacovigilance database. [in French] Therpaie 2001; 56:51–5.
12. Bhala N., Plevris JN. Portal hypertensive enteropathy; an usual cause of gastrointestinal bleeding in cirrhosis: or is it? JR Coll Physicians, 2006; 36; 208 – 210.
13. Bjorkman D. Nonsteroidal anti–inflammatory drug associated toxicity of the liver, lower gastrointestinal tract and the esophagus. Am J Med 1998; 105:17–21S.
14. Bort R, Ponsoda X, Jover R, et al. Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther 1999; 288:65–72.
15. Danan G, Benichou Ch. Causality assessment of adverse reactions to drugs – I. A novel method based on the conclusions of international consensus meetings: applications to drug–induced liver injury. J. Clin Epidemiol, 1993; 4: 111–4.
16. Dierkes–Globisch A, Schafer R, Mohr HH. Asymptomatic diclofenac–induced acute hepatitis [in German]. Dtsch Med Wochenschr 2000; 125:797–800.
17. Fontana RJ, McCashland TM, Benner KG, et al. Acute liver failure associated with prolonged use of bromfenac leading to liver transplantation. The Acute Liver Failure Study Group. Liver Transpl Surg 1999; 5:480–4.
18. Garcia Rodriguez LA, Perez Guttan. The role of non–steroidal anti–inflammatory drugs in acute liver injury. BMJ, 1994; 6: 107–17.
19. Gastritis and Other Gastropathies. In: Feldman, M.; Friedman, L.S.; Sleisenger, M.H. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology/Diagnosis/Management. 7th ed. [2–volume set]. St. Louis, MO: Saunders. 2002. p. 810–827.
20. Greaves RR, Agarwal A, Patch D, et al. Inadvertent diclofenac rechallenge from generic and non–generic prescribing, leading to liver transplantation for fulminant liver failure. Eur J Gastroenterol Hepatol 2001; 13:71–3.
21. Lacroix I, Lapeyre–Mestre M, Bagheri H. Nonsteroidal anti–inflammatory drug–induced liver injury: a case–control study in primary care. Fundamental & Clinical Pharmacology, 2004; 18: 201–6.
22. Lapeyre–Mestre M, Rueda de Castro AM. Non–steroidal anti–inflammatory drug–related hepatic damage in France and Spain. Fundamental & Clinical Pharmacology, 2006; 20: 391–5.
23. Mabee CL, Mabee SW, Baker PB. Fulminant hepatic failure associated with etodolac use. Am J Gastroenterol, 1995; 90: 659–61.
24. Maddrey WC, Maurath CJ, Verburg KM, et al. The hepatic safety and tolerability of the novel cyclo–oxygenase–2 inhibitor celecoxib. Am J Ther 2000; 7:153–8.
25. Martin R, Biswas P. The incidence of adverse events and risk factors for upper gastriointestinal disorders associated with meloxicam. Br J Clin Pharmacol, 2000; 50: 35–42.
26. Masubuchi Y, Horie T. A possible mechanisms of naproxen–induced ipid–peroxidation in liver microsomes. Pharmacol Toxicol, 2001; 89: 43–8.
27. Masubuchi Y, Horie T. Structural requirements for the hepatotoxicity of nonsteroidal anti–inflammatory drugs. J Pharmacol Exp Ther, 1998: 287: 208–13.
28. Masubuchi Y, Nakayama S, Horie T. Roe of mitochondrial permeability transition in diclofenac–induced hepatotoxicity. Hepatology, 2002; 35: 544–51.
29. Masubuchi Y, Nakayama S, Horie T. Role of mitochondrial permeability transition in diclofenac–induced hepatotoxicity in rats. Hepatology 2002; 35:544–551.
30. Masubuchi Y, Yamada S, Horie T. Possible mechanisms of hepatocyte injury induced by diphenylamine and its structurally related NSAIDs. J Pharmacol Exp Ther, 2000; 292: 982–7.
31. Mc Cormick PA, Kennedy F, Curry M. COX 2 inhibitor and fulminant hepatic failure. Lancet, 1999; 353: 40–1.
32. McCormack TT, Sims J, Eyre–Brook I, et al. Lesions in portal hypertension: inflammatory gastritis or congestive gastropathy? Gut 1985;26:1226–32.
33. Merlani G, Fox M, Oehen HP, Cathomas G, Renner EL, Fattinger K, Schneeman M, Kullak–Ublick GA. Fatal hepatotoxicity secondary to nimesulide. Eur J Clin Pharmacol 2001; 57:321–6.
34. O’Connor N, Dargan PI, Jones AL. Hepatocellular damage from non–steroidal anti–inflammatory drugs. Q J Med, 2003: 96: 787–791.
35. Ponsoda X, Pareja E, Gomez–Lechon MJ, et al. Drug biotransformation by human hepatocytes. In vitro/in vivo metabolism by cells from the same donor. J Hepatol 2001; 34:19–24.
36. Purdum PP 3rd, Shelden SL, Boyd JW, Shiffman ML. Oxaprozin–induced fulminant hepatitis. Ann Pharmacother 1994; 28:1159–61.
37. Rabinovitz M, Van Thiel DH. Hepatotoxicity of non–steroidal anti–inflammatory drugs. Am J Gastroenterol 1992; 87: 1696–704.
38. Rabkin JM, Smith MJ, Orloff SL, et al. Fatal fulminant hepatitis associated with bromofenac use. Ann Pharmacother 1999; 33:945–7.
39. Rabkin JM, Smith MJ, Orloff SL. Fatal fulminant hepatitis associated with bromfenac use. Ann Pharmacother, 1999; 33: 945–7.
40. Rainsford KD. An analysis from clinico–epidemiological data of the principal adverse events from the COX–2 selective NSAID, nimesuide, with particular reference of hepatic injury. Inflammopharmacology, 1998; 6: 203–21.
41. Riley TR, Smith JP. Ibuprofen–induced hepatotoxicity in patients with chronic hepatitis C: A case series. Am J Gastroenterol 1998; 93:1563–5.
42. Rodriguez–Gonzalez FJ, Montero JL, Puente J, et al. Orthotopic liver transplantation after subacute liver failure induced by therapeutic doses of ibuprofen. Am J Gastroenterol 2002; 97:2476–7.
43. Rubenstein J., Laine L. Systematic review: the hepatotoxicity of non–steroidal anti–inflammatory drugs. Aliment Pharmacol Ther, 2004; 20: 373–80.
44. Samonakis DN, Triantos CK, Thalheimer U: Management of portal hypertension. Postgrad Med J 2004 Nov; 80(949): 634–41.
45. Schattner A, Sokolovskaya N. 2000
46. Sgro C, Clinard F, Quazir K. Incidence of drug–induced hepatic injuries. Hepatology, 2002; 36: 451–5.
47. Sherlock S, Dooley J, eds: Diseases of the Liver and Biliary System. Oxford, UK: Blackwell Science; 1997.
48. Tan H, Ong WM, Lai SH. Nimesulide–induced hepatotoxicity and fatal hepatic failure. Singapore Med J, 2007; 48(6): 582–5.
49. Teoh NC, Farrell GC. Hepatotoxicity associated with non–steroidal anti–inflammatory drugs. Clin Liver Dis, 2003; 7: 401–13.
50. Traversa G, Bianchi C, DaCas R. Cohort study of hepatotoxicity associated with nimesulide and other non–steroidal anti–inflammatory drugs. BMJ, 2003; 327: 18–22.
51. Wongcharatrawee S, Groszmann RJ: Diagnosing portal hypertension. Baillieres Best Pract Res Clin Gastroenterol 2000 Dec; 14(6): 881–94.
Комментариев нет:
Отправить комментарий